The introduction of stents in the treatment of coronary artery disease has dramatically altered the practice of coronary artery bypass grafting surgery. New complications caused by percutaneous coronary interventions (PCI) with the implantation of stents has emerged and affected the result when surgery is subsequently required.
The stent was initially utilized for the treatment of complications associated with percutaneous coronary angioplasty, specifically acute occlusion and restenosis. Employed as support, an expanded metallic mesh impedes the phenomenon of 'recoil', one of the factors responsible for genesis of post-angioplasty restenosis.
However, recent evidence has demonstrated that the coronary stent may induce the appearance of Systemic Inflammatory Response Syndrome (SIRS) [1,2], where restenosis after stent implantation constitutes the earliest manifestation of an inflammatory reaction. SIRS, the consequent endothelial dysfunction and ischemia are events that follow and are little understood.
The inflammatory reaction triggered by the insertion of the stent is caused and maintained by the following factors:
1 - expansion of the stent with rupture of the atherosclerotic plaque and the tunica media
2 - maintenance of the radial pressure of the stent on the arterial wall
3 - the presence of a metallic foreign body
4 - ischemic phenomenon induced by endothelial dysfunction
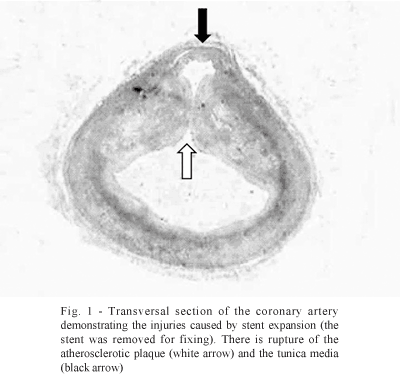
The implantation and expansion of the stent determines a coronary vascular injury that is very much more severe to the lesion produced by balloon angioplasty. This follows the concept of "the bigger the better", initially suggested by Kuntz et al. [3] where the high dilation pressure [4], in which the expansion of the stent creates the greatest luminal diameter possible, causes severe structural alterations to the wall of the coronary artery. Figure 1 demonstrates the severity of the lesion caused on the arterial wall by the expansion of the stent with the rupture of the atherosclerotic plaque and the tunica media.
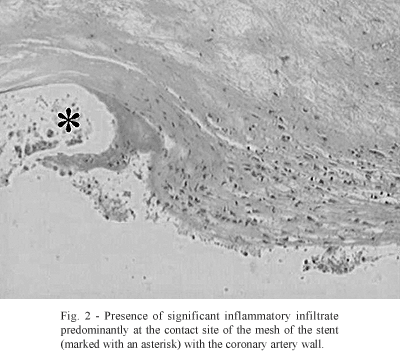
Maintenance of the radial pressure of the stent to prevent 'recoil' acts as a factor of aggression, stimulating the inflammatory response. It has been demonstrated that the initial inflammatory reaction is more accentuated at the points of greatest pressure of the metallic structure on the arterial wall, as can be seen in figure 2.
The presence of the intra-coronary metallic mesh is as a foreign body, causing the appearance of a granuloma-type continuous inflammatory response. Finally, the endothelial dysfunction consequent to the inflammatory response induces thrombosis and ischemic phenomenons, with the inhibition of vasodilator factor synthesis (in particular nitric oxide) and increases in the liberation of endothelin 1, a potent vasoconstrictor.
There are no doubts, nowadays, that atherosclerotic disease has an inflammatory origin. Mechanical injuries of the arteries or exposure to atherogenic stimuli induce an inflammatory response, with adherence and migration of leukocytes to the vessel wall creating macrophages (which form foam cells). There is liberation of growth factors and cytokines with recruitment of smooth muscle cells and stimulation of neo-intimal proliferation, leading to the accumulation of lipids and endothelial dysfunction [5].
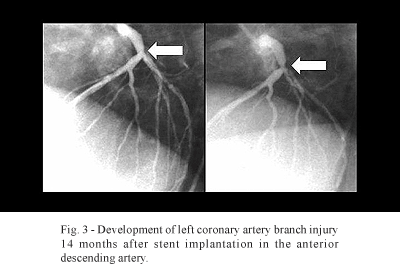
The insertion of a stent acts in synergy with the atherosclerotic plaque, increasing the inflammatory response that already exists in the coronary arterial wall. As a consequence, there may be acceleration in the evolution of atherosclerotic disease. Figure 3 illustrates a typical example of accelerated progression of coronary disease, with the development of injury of the left coronary branch in a patient 14 months after stent implantation.
Recent studies show that inflammatory markers increase in the blood after stent implantation. Almagor et al. [1] demonstrated that levels of reactive protein C in patients after coronary stent implantation were persistently high over the medium term, constituting a continuous systemic inflammatory response.
Another study demonstrated that there is long-term persistence of the high levels of sanguineous inflammatory mediators in patients submitted to stent implantation. Serous levels of the soluble receptors of interleukin-2 (sIL-2R) for activation of T-lymphocytes (markers of immunity mediated by cells) are still high four months after stent implantation. The pro-inflammatory cytokines and the acute phase proteins are also precociously liberated in the peripheral circulation and immunity mediated by cells persists for an indefinite time after stent implantation [6].
Navarro-Lopez et al. [7] demonstrated that at 6 months of evolution, patients with stent restenosis presented with an increase in the inflammatory activity expressed by a rise of the cytotoxic T lymphocytes CD3+/CD56+ and activated monocytes CD11b.
These findings of persistently high inflammatory markers confirm the existence of continuous SIRS after the implantation of coronary stents.
Some diseases are recognized by their evolution with a pattern of persistently high inflammatory markers. The prognosis is still not known, but these patients have a tendency to present with recurrent infections and an increase in the incidence of osteo-articular events.
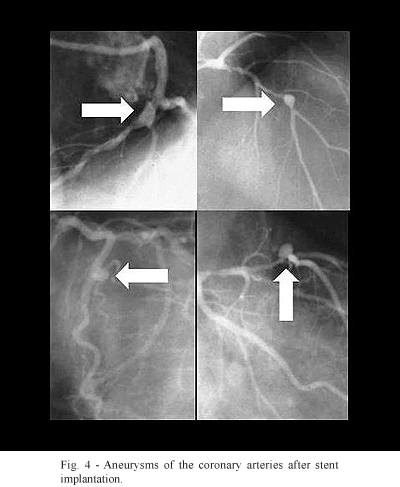
Mechanical injury and subsequent inflammation of the coronary arterial wall produce the appearance of weak zones, causing the formation of aneurysms, as can be seen in Figure 4. Evolution and prognosis of these aneurysms of the coronary artery are little studied, but published reports confirm that they can rupture and cause a fatal outcome [8].
As an additional factor of the inflammatory reaction to PCI with stents, there are the acute and chronic ischemic processes. Acute ischemia is caused by two factors in relation to stent implantation. The first is by atheroembolism following the expansion of the stent with rupture of the atherosclerotic plaque and liberation of its contents, with the creation of numerous emboli that occlude the distal coronary microcirculation. The other mechanism involved is the obstruction of the lateral branches of the coronary artery in the region of the stent implantation site, with localized infarctions of the regions supplied by these branches. Cantor et al. [9], studying the incidence and clinical significance of augmented cardiac troponin I (cTnI) after PCI, demonstrated that 48% of the patients had on increase in cTnI levels after PCI, compatible to acute myocardial infarction. This increase of cTnI was associated with a significant increase in the risk of death or infarction and worse clinical outcomes in the first 90 days after the procedure.
Chronic ischemia occurs following deterioration of the endothelial function, owing to persistence of the coronary inflammatory response. It has been demonstrated that there is an inhibition of the nitric oxide synthesis and liberation of vasoconstrictor factors (mainly endothelin-I) [10]. Consequently there is loss of the vasodilation capacity of the coronary artery and ensuing prejudice to the offer-demand linking mechanism of oxygen for the cardiac muscle. This results in sequential situations of myocardial ischemia due to the impossibility of increasing the coronary blood flow in situations of increased demand of the cardiac muscle. Also this explains the situations that patients with stents present with angina or myocardial infarction with the stent apparently patent, as evidenced by cinecoronariography.
Furthermore, there is evidence of systemic repercussions of endothelial dysfunction. Wu et al. [11] investigated systemic endothelial function in forearm arteries of patients after stent implantation. In patients with angiographic restenosis of the stent, reactive hyperemia of the forearm was seen to be worse when compared with control individuals and this was associated with liberation of endothelin-I, which was elevated in the coronary circulation immediately after PCI.
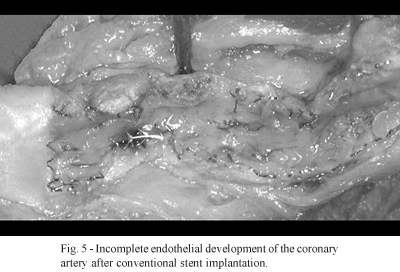
The mechanism of thrombosis might still be aggravated by exposure to the metallic structure of the stent in the lumen of the artery, as, frequently, endothelial development surrounding the metallic parts remains incomplete (Figure 5).
Clopidogrel, a platelet anti-aggregate, has been extensively used to attempt to control the thrombotic process. However, recent studies show that antithrombotic protection of clopidogrel is incomplete, exhibiting a considerable individual heterogeneity, with some individuals demonstrating potent antiplatelet inhibition and others a weak response [12]. Even more recently, another complicating factor arose linked to the utilization of clopidogrel. This drug is capable of inducing SIRS, as was demonstrated by Wolf et al. [13]. Is this another factor that acts in synergy with the inflammatory reaction of atherosclerotic disease and with an inflammatory response induced by the insertion of the stent, increasing even more the expression of SIRS in patients?
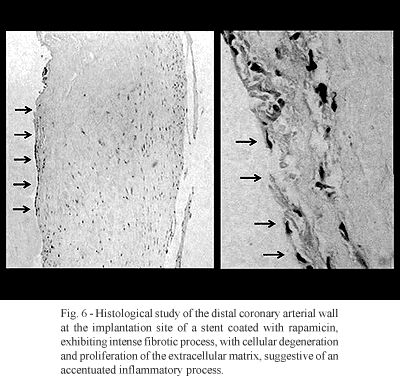
Finally, the introduction of technology of stents coated with drugs (DES-drug-eluding stents) increased another variable interacting with the arterial wall. These drugs possess effects that lead to the appearance of other specific complications (idiosyncrasy). Examples are the recent launch of stents with rapamicin (sirolimus) (Cypher TM) and paclitaxel (Taxus TM). Despite of the antiproliferative effect of sirolimus (rapamicin) apparently manifested and a reduction of the endothelial growth and intra-stent restenosis, the inflammatory response is aggravated at the ends of the stent, producing an effect of increasing the inflammatory response and intra-segmental restenosis [14] (figure 6).
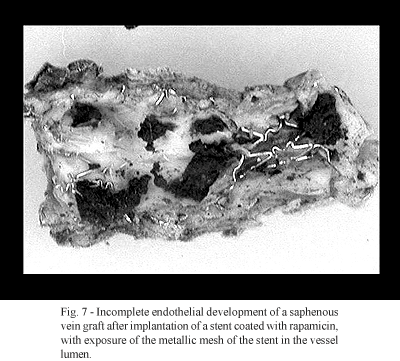
Sirolimus also has been associated with the appearance of reactions of hypersensitivity to medicines, such as pain, exanthema and cutaneous eruptions, respiratory alterations, urticaria, pruritus, fever and blood pressure alterations [15]. The long-term effects of these complications are unknown, Furthermore, sirolimus has been demonstrated as a platelet agonist, stimulating platelet aggregation and inducing intra-coronary thrombosis [16]. This complication is currently being investigated by the Food and Drug Administration in the USA [15].
Other stents coated with drugs will probably exhibit specific side effects, forcing cardiac surgeons and clinical cardiologists to employ specific interventions for each type of device [17].
As suggested, the most obvious implication is that probably the coronary artery bypass grafting surgery may not provide patients with previously implanted stents the same results as those results obtained in patients without stents, results published over the last decades. The consequences of chronic inflammation of the coronary artery (arteritis) and myocardium (myocarditis) are still unknown, as are how these factors can affect the prognosis of the patient. Only future research in this area might elucidate all these questions.
BIBLIOGRAPHIC REFERENCES
1. Almagor M, Keren A, Banai S. Increased C-reactive protein level after coronary stent implantation in patients with stable coronary artery disease. Am Heart J 2003;145:248-53.
2. Gomes WJ, Giannotti-Filho O, Catani R, Paez RP, Hossne NA Jr., Buffolo E. Alterações inflamatórias das artérias coronárias e do miocárdio induzidas por stents coronários. Rev Bras Cir Cardiovasc 2002;17:293-8.
3. Kuntz RE, Safian RD, Carrozza JP, Fishman RF, Mansour M, Baim DS. The importance of acute luminal diameter in determining restenosis after coronary atherectomy or stenting. Circulation 1992;86:1827-35.
4. Colombo A, Hall P, Nakamura S, Maiello L, Martini G, Gaglione A et al. Intracoronary stenting without anticoagulation accomplished with intravascular ultrasound guidance. Circulation 1995;91:1676-88.
5. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002;105: 1135-43.
6. Caixeta AM, Brito Jr FS, Serrano CV, Diniz C, Brasileiro AL, Tura B et al. Cytokines and inflammatory markers are released early after coronary stenting and are related to 6-month clinical outcome. Eur Heart J 2002;23(suppl):555.
7. Navarro-López F, Francino A, Serra A, Enjuto M, Reverter JC, Jimenez de Anta T et al. Late L-lymphocyte and monocyte activation in coronary restenosis. Evidence for a persistent inflammatory/immune mechanism? Rev Esp Cardiol 2003;56:465-72.
8. Beach L, Burke A, Radentz S, Virmani R. Spontaneous fatal rupture of a coronary arterial aneurysm into the right ventricle.Am J Cardiol 2001;88:99-100.
9. Cantor WJ, Newby LK, Christenson RH, Tuttle RH, Hasselblad V, Armstrong PW et al. Prognostic significance of elevated troponin I after percutaneous coronary intervention. J Am Coll Cardiol 2002;39:1738-44.
10. Hafizi S, Allen SP, Goodwin AT, Chester AH, Yacoub MH. Endothelin-1 stimulates proliferation of human coronary smooth muscle cells via the ET(A) receptor and is co-mitogenic with growth factors. Atherosclerosis 1999;146:351-9.
11. Wu TC, Chen YH, Chen JW, Chen LC, Lin SJ, Ding PY et al. Impaired forearm reactive hyperemia is related to late restenosis after coronary stenting. Am J Cardiol 2000;85:1071-6.
12. Jaremo P, Lindahl TL, Fransson SG, Richter A. Individual variations of platelet inhibition after loading doses of clopidogrel. J Intern Med 2002;252:233-8.
13. Wolf I, Mouallem M, Rath S, Farfel Z. Clopidogrel-induced systemic inflammatory response syndrome. Mayo Clin Proc 2003;78:618-20.
14. Teirstein PS, King S. Vascular radiation in a drug-eluting stent world: it's not over till it's over. Circulation 2003;108:384-5.
15. FDA Public Health Web Notification. Information for physicians on sub-acute thromboses (SAT) and hypersensitivity reactions with use of the Cordis CYPHERT coronary stent. Issued 10/29/2003. Available from: URL:http://www.fda.gov/cdrh/safety/cypher.html.
16. Choi SB. CYPHER coronary stents and risk of thrombosis [Editorial]. CMAJ 2003;169:218.
17. Gomes WJ, Giannotti-Filho O, Paez RP, Hossne NA Jr., Catani R, Buffolo E. Coronary artery and myocardial inflammatory reaction induced by intracoronary stent. Ann Thorac Surg 2003;76: 1528-32.
 All scientific articles published at www.rbccv.org.br are licensed under a Creative Commons license
All scientific articles published at www.rbccv.org.br are licensed under a Creative Commons license













 English PDF
English PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket