Primary cardiac myxosarcoma is a very rare disease and is difficult to differentiate from myxoma, both clinically and pathologically. In this study, the authors report the first case of surgical excision of left atrial myxosarcoma in Brazil, in a 36-year-old woman. The operation was sucessful, and the patient remains asymptomatic for more than 180 postoperative days (Functional Class I - NYHA), with no signs of relapse of the tumor.
O mixossarcoma é uma forma rara de neoplasia cardíaca primária de difícil diferenciação clínica e patológica com o mixoma. Até onde os autores têm conhecimento, este é o primeiro relato de caso na literatura nacional indexada com o tratamento cirúrgico do mixossarcoma atrial esquerdo, em paciente do sexo feminino, de 36 anos de idade, cuja evolução pós-operatória tem sido satisfatória, encontrando-se em classe funcional I (NYHA) e em remissão do processo tumoral há mais de 180 dias.
Introduction
Cardiac myxosarcoma is a rare form of primary malignant neoplasia which is difficult to differentiate both clinically and pathologically from myxoma [1]. The term 'myxosarcoma' is not commonly used in the classification of tissue tumors, but restricted to heart tumors which are myxoid but do not have diagnoses similar to other sarcomas [2].
Histologically, the tumor appears like a neoplasia composed of undifferentiated cells originating from the endocardium. The malignant cells show areas of pleomorphism and hyperchromatism with intense mitotic activity [3].
Here we report on the first case of successful treatment of this rare primary cardiac neoplasia described in Portuguese.
Case Report
We report on the case of a 36-year-old female patient with a history of transitory ischemic events since the age of 19 years old. In the last episode (8 months before admission) she remained in a coma for 4 days. A neurological examination by magnetic nuclear resonance excluded cerebral arteriovenous malformations and expansive processes of the brain. The patient evolved with motor deficiencies on the right and aphasia.
She was admitted to hospital without cardiovascular symptoms. The patient was in a good general state, conscious, eupneic, acyanotic, with normal color, anicteric and afebrile. The neurological examination demonstrated aphasia with predominance of expression, central to right facial paralysis and paresis of the right side with brachial predominance.
Cardiovascular examinations showed a regular heartbeat with normal heart sounds and without murmurs. The heart frequency was 75 bpm and the blood pressure was 110/70 mmHg.
A chest radiograph showed that the pulmonary fields were clear with a normal cardiac area. The electrocardiogram demonstrated sinus rhythm without evidence of overload or hypertrophy.
A transthoracic echocardiogram evidenced, in the left atrium (LA) a heterogeneous echogenic mass measuring approximately 3.09 x 1.91 cm with great mobility but adhered to the upper portion of the interatrial septum.
A transesophageal echocardiogram demonstrated a 'spongiform' mass in the LA measuring around 4 x 3.2 cm with much mobility and apparently adhered to the upper portion of the interatrial septum. It was a heterogeneous echogenic mass, entering the left ventricle during diastole.
Cardiac catheterism showed the left ventricle had the diastolic volume and contractile function conserved. The LA was slightly enlarged and emptying without difficulty.
Thus, a cardioembolic event was diagnosed, whose probable etiology was left atrial myxoma. Faced with the clinical state of the patient and the complementary examinations, a surgical intervention was indicated.
A median sternotomy was performed. Conventional cardiopulmonary bypass was established with slight hypothermia at 34 ºC. In anoxic arrest with single aortic clamping during 10 minutes, a left atriotomy was performed allowing complete excision of the tumoral mass. The mass was fixed at the base of the posterior mitral leaflet, practically free, without evident anchoring points on the atrial structures. The surgical procedure was concluded in the normal manner. The postoperative period was uneventful. The patient was released from hospital five days after the surgery.
During the outpatient follow-up of 180 days, the patient was completely free of symptoms in functional class I (NYHA) and without evidence of tumor seen by echocardiography.
This current report was approved by the Research Ethics Committee of University Hospital of the Federal University of Maranhão.
Histopathologic analysis
By macroscopy, the tumor appeared as a gelatinous friable brownish mass of 4 x 2 x 1 cm (Figure 1). By microscopy, some bizarre necro-hemorrhagic, myxoid and hyperemia cells were observed (Figure 2).

Fig. 1 - Macroscopic appearance of left atrial myxosarcoma
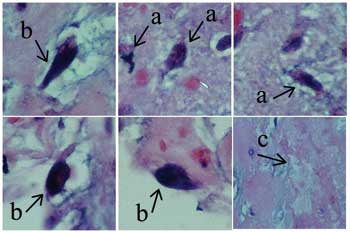
Fig. 2 - Microphotography at 400 x showing atypical mitosis (a) bizarre cells (b) and area of necrosis (c)
Primary cardiac neoplasms are seen in from 0.002 to 0.3% of cases in autopsies, of which 30% are myxomas and 20-30% are malignant, almost always sarcomas. Hence, the incidence of malignant primary cardiac tumors varies between 0.0004% and 0.025%, among which myxosarcomas represent less than 1% [1,3].
The rarity of this tumor does not allow an exact evaluation of its clinical course or its treatment. Until 1949, there were 6 published cases of myxosarcoma. In 1985 a case was reported in the Mayo clinic and in the following year another in the Texas Heart Institute [3,4]. In 1992, Burke et al. [2] described 3 cases and in 2001, the first case of cardiac myxosarcoma was reported in Korea [1]. In our review of the literature of the main databases (PubMed, LILACS) in the period from 1966 to 2005, there were no reports in Portuguese about this type of tumor.
The origin of cardiac myxosarcoma remains unclear, the fact that myxosarcoma corresponds to the malignant transformation of cardiac myxoma is controversial as there is no solid evidence to confirm this transformation [1]. There are three reasons that refute this hypothesis. Firstly, compound tumors formed by sarcoma and myxoma probably do not exist and the tendency for recurrence of myxoma depends on hereditary factors and not on histological or atypical appearance. Secondly, with the exception of angiosarcoma, the majority of sarcomas, including myxosarcoma, grow in the LV. And finally, the myxoid substrate can occur in all types of cardiac sarcomas as a consequence of the intracavitary location and not as a histogenesis reflex [2,5].
The most important criteria in the identification of myxosarcoma are the absence of typical chordae, rings and capillary structures formed by the myxomatous cells. The degree of cellularity of myxosarcoma is variable, although myxoma, can occasionally be very cellularized; however, there are inevitable focuses of atypical hyperchromatic cells in myxosarcoma [5].
The histological material of the current case showed areas of necrosis and cellular indifferentiation, as well as differentiated areas which resembled benign atrial myxoma. However, the cells were clearly malignant showing areas of pleomorphism and hyperchromicity, with intense mitotic activity, consistent with findings of myxosarcoma described in the literature [1-6].
Despite the histological finding of necrosis and the mitotic rate being greater than 10 x 10 per field of high magnification constituting a factor of poor prognosis [2], the patient is in complete remission of the neoplastic process 180 days after resection of the tumor.
In the current study we report on a case of prolonged clinical evolution, nevertheless, we did not organize the clinical and histopathologic elements to infer about the malignancy of a primary myxoma. A report of a slow and progressively developing myxosarcoma has already been published by Roh et al. [1]. These authors published a case where the patient presented with systemic metastasis at admission; even so the patient had a satisfactory postoperative evolution, with complete regression of the primary and metastatic tumoral process over 37 months of follow up.
Until now, there are no imaging techniques capable of determining the definitive diagnosis of these tumors. Only using endomyocardial biopsy and thoracotomy it is possible to characterize in vivo these cardiac masses.
Immediate histological analysis is essential to confirm the diagnosis and establish the most adequate surgical treatment, which in our experience, has proved to be effective in a follow up of six months.
In conclusion, atrial myxosarcoma is a rare and distinct entity that requires a high degree of clinical suspicion for its diagnosis, as the symptoms may be vague as we reported here.
REFERENCES
1. Roh MS, Huh GY, Jeong JS, Lee GD, Hong SH. Left atrial myxosarcoma with systemic metastasis: a case report. J Korean Med Sci. 2001;16(1):111-4.
2. Burke AP, Cowan D, Virmani R. Primary sarcomas of the heart. Cancer. 1992;69(2):387-95.
3. Harris GJ, Tio FO, Grover FL. Primary left atrial myxosarcoma. Ann Thorac Surg. 1993;56(3):564-6.
4. Vander Salm TJ. Unusual primary tumors of the heart. Semin Thorac Cardiovasc Surg. 2000;12(2):89-100.
5. Morin JE, Rahal DP, Hüttner I. Myxoid leiomyosarcoma of the left atrium: a rare malignancy of the heart and its comparison with atrial myxoma. Can J Cardiol. 2001;17(3):331-6.
6. Samal AK, Ventura HO, Berman A, Okereke C, Gilliland YE, Willis GW. Myxosarcoma: a rare primary cardiac tumor. J La State Med Soc. 2002;154(6):308-12.
Note of the editor
There were differences between the opinions of the referees in respect to the anatomopathological findings reported. We accepted in good faith, additional comments in reference to this article that stimulate doubts, opening the possibility of discussion that may be fertile and instructive.
 All scientific articles published at www.rbccv.org.br are licensed under a Creative Commons license
All scientific articles published at www.rbccv.org.br are licensed under a Creative Commons license













 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket